Research Interests
Research Interests
DNA Double-Strand Break Repair
DNA Double-Strand Break Repair
A primary interest of the group is to help in the battle against breast cancer by providing an understanding at the atomic level of the macromolecules and complexes involved in the progression of the disease. Current targets include Replication Protein A (RPA), Rad51, and Rad52.
RPA is a single-stranded DNA binding protein and a key player in general DNA metabolism. RPA was first broadly recognized as being essential for DNA synthesis and has since been shown to be intrinsic to DNA recombination, repair, and transcription. RPA enjoys such a critical role through its interactions with other proteins. For example, a protein-protein complex formed by RPA and repair factor XPA recognizes damaged DNA in the first step of nucleotide excision repair. Furthermore, interactions between RPA and repair factor Rad52 are essential to homologous recombination.
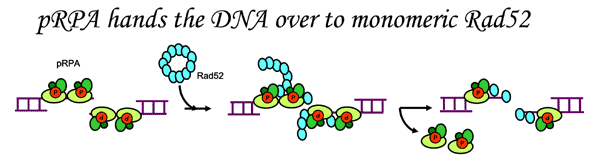
Rad52 was first broadly recognized for its importance in yeast survival when treated with ionizing radiation. Rad52 forms a ring structure, bonds to DNA double-strand breaks, and interacts with RPA and repair factor Rad51. Our data indicated phosphorylated RPA binds to monomeric Rad52 causing strand transfer (ACS link). Familial breast cancer genes, BRCA1 and BRCA2 are also involved in this double-strand break repair pathway.
MnSOD (μg Cystallization and Neutron Diffraction)
MnSOD (μg and N0)
Ionizing radiation in space is a very prominent health hazard to astronauts and is deadly during prolonged space travel, such as planned trips to Mars. Radiation is a significant source of reactive oxygen species (ROS), which cause major health problems even for the average person on Earth. Excessive ROS create oxidative stress, which is involved in the etiology of cancer, neurological disorders, heart disease, etc.
Superoxide dismutases (SODs) are enzymes that provide the first line of defense against ROS. SODs scavenge two superoxide molecules and allow conversion into regular diatomic oxygen and hydrogen peroxide through a cyclic redox fashion utilizing a catalytic metal ligand. Despite the biological importance of SODs, the complete enzymatic reaction is unknown due to limitations in identifying the binding sites of the substrate, hydrogens, and products.
Structural data of complexes and intermediates can be identified by growing large crystals of perdeuterated protein (>1 mm3) in microgravity (μg) on the International Space Station and solving the structures using neutron and X-ray diffraction.
Results of this work can be found here.
Modulated Crystallography
Modulated Crystallography
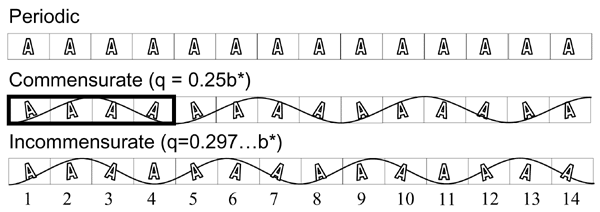
For a normal periodic crystal, the X-ray diffraction pattern can be described by an orientation matrix and a set of three integers that indicate the reciprocal lattice points. Those integers determine the spacing along the reciprocal lattice directions. In aperiodic crystals, the diffraction pattern is modulated and the standard periodic main reflections are surrounded by satellite reflections. We have successfully indexed and refined the main unit cell and q-vector of an incommensurately modulated, aperiodic protein crystal of a profilin:actin complex. The indexing showed that the modulation is along the b direction in the crystal, which corresponds to an 'actin ribbon' formed by the crystal lattice. Interestingly, the transition to the aperiodic state has been shown to be reversible and the diffraction pattern returned to the periodic state during data collection. It is likely that the protein underwent a conformational change that affected the neighboring profilin:actin molecules in such a way as to produce the observed modulation in the diffraction pattern. We have also cross-linked and cryo-trapped the incommensurately modulated crystal state.
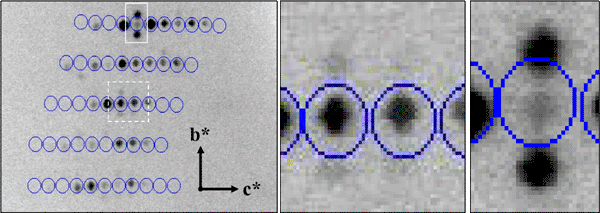
Modulated Profilin:Actin Diffraction Pattern (main reflections circled)
Crystal Perfection
Crystal Perfection
The group is also very interested in developing methods for the quantitative comparison of crystal quality. We employ two approaches.
One experimental method involves using highly-parallel, highly monochromatic synchrotron radiation, super-fine phi slicing and a CCD area detector to simultaneously measure mosaicity and diffraction resolution from hundreds of reflections at a time. These measurements can be used to quantitatively compare protein crystals grown from different methods, for example earth gravity vs microgravity growth or the effects of impurities. We are continuing to develop these crystal quality methods and have written a GUI program for data processing called BEAM-ish.
The second experimental method is to use digital topography to analyze internal crystal structure. A program called Ripple has been developed to collect and analyze this data.
Results of this work can be found here.

